To demonstrate the utility of The M1CR0B1AL1Z3R web server, we provide detailed analysis of three different databases:
Of note, these examples highlight the main usage of the web server: a comparative analysis of assembled microbial genomes. It is not intended for raw metagenomics data (i.e., before assembly and binning). The results for each dataset are provided below.
[1] M1CR0B1AL1Z3R analysis of different isolates from the same E. coli strain. This dataset is comprised of 50 pathogenic Escherichia coli lineage ST131 genomes (McNally A, et al., 2016). This dataset represents highly similar clinical isolates of a specific bacterial species. Several isolates were not fully assembled and contained dozens and even hundreds of contigs. This demonstrates the capability of M1CR0B1AL1Z3R to handle partially assembled genomes. We added an outgroup sequence to this dataset, the genomic sequence of Escherichia fergusonii. The results for this example can be found here.
[1a] The identification of ORFs in each genome is one of the first computation steps. Below is a violin plot representing the dispersion of the ORF count per genome.
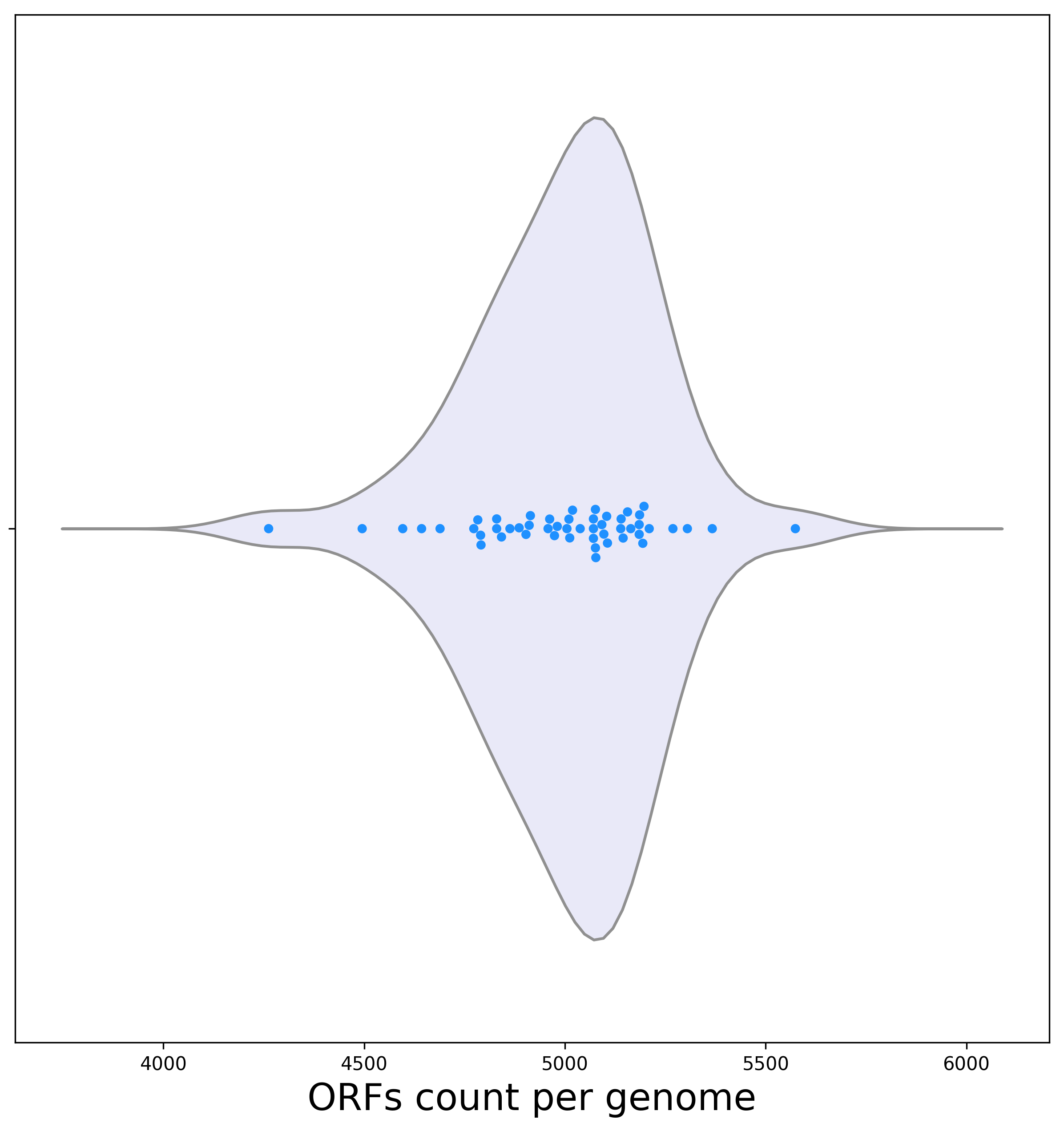
Biological interpretation: As can be seen, there is high variation in the number of encoded ORFs. This variation is often attributed to the high rate of horizontal gene transfer. However, it is also possible that some variance may result from missing data in some genomes. This plot can thus provide a gateway for further research concerning the source of differences in the number of ORFs among genomes. The genome with the lowest number of ORFs is E. fergusonii ATCC 35469. It encodes 4,262 ORFs. The next smallest genome in terms of ORF count encodes 4,495 ORFs (genome J09).
[1b] An additional analysis that is conducted before searching for orthologous sequences is the determination of the distribution of the GC composition (the relative proportion of the G and C nucleotides out of the total number of nucleotides in protein coding sequences). Below is a violin plot representing the dispersion of the GC among genomes.
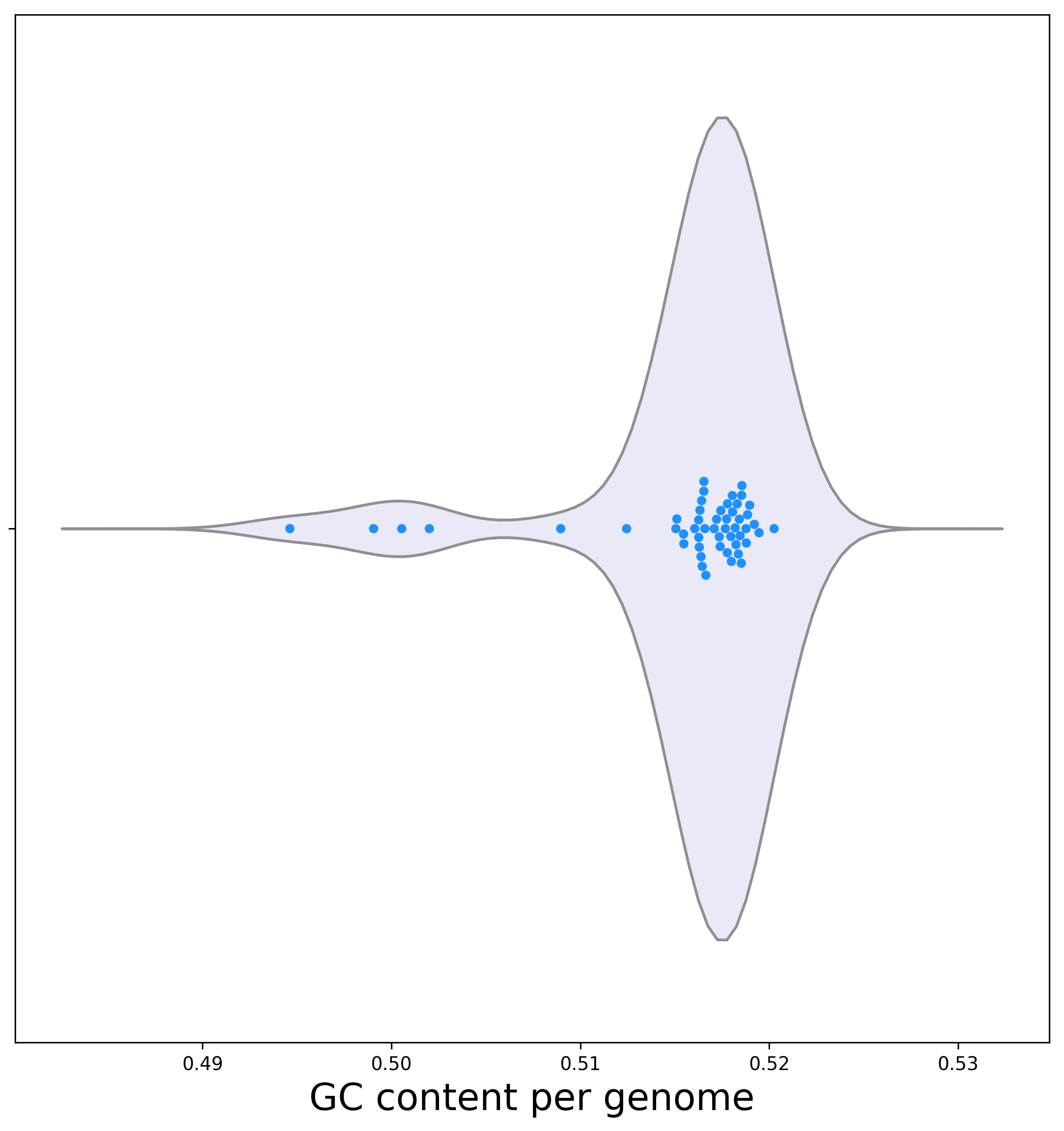
Biological interpretation: the vast majority of genomes have a GC content in the range of 51% to 52%. Few variants show atypical GC content. The names of the isolates with a lower GC content can be extracted from the provided output file. For example, the isolate with the lowest GC content is UTI423. It could be interesting to further study whether a correlation exists between isolates with the atypical GC content and those with extreme number of ORFs, or if they are relatively diverged from the other isolates on the phylogenetic tree. Testing this, we found that the two genomes (UTI423 and UTI587) with the lowest GC content (less than 50%) had the highest number of ORFs. This can be explained by extensive recent events of horizontal gene transfer acquired from AT rich donors. Of note, horizontally transferred genes are often AT rich, further supporting our biological interpretation (Daubin V, et al., 2003).
[1c] Orthologs detection
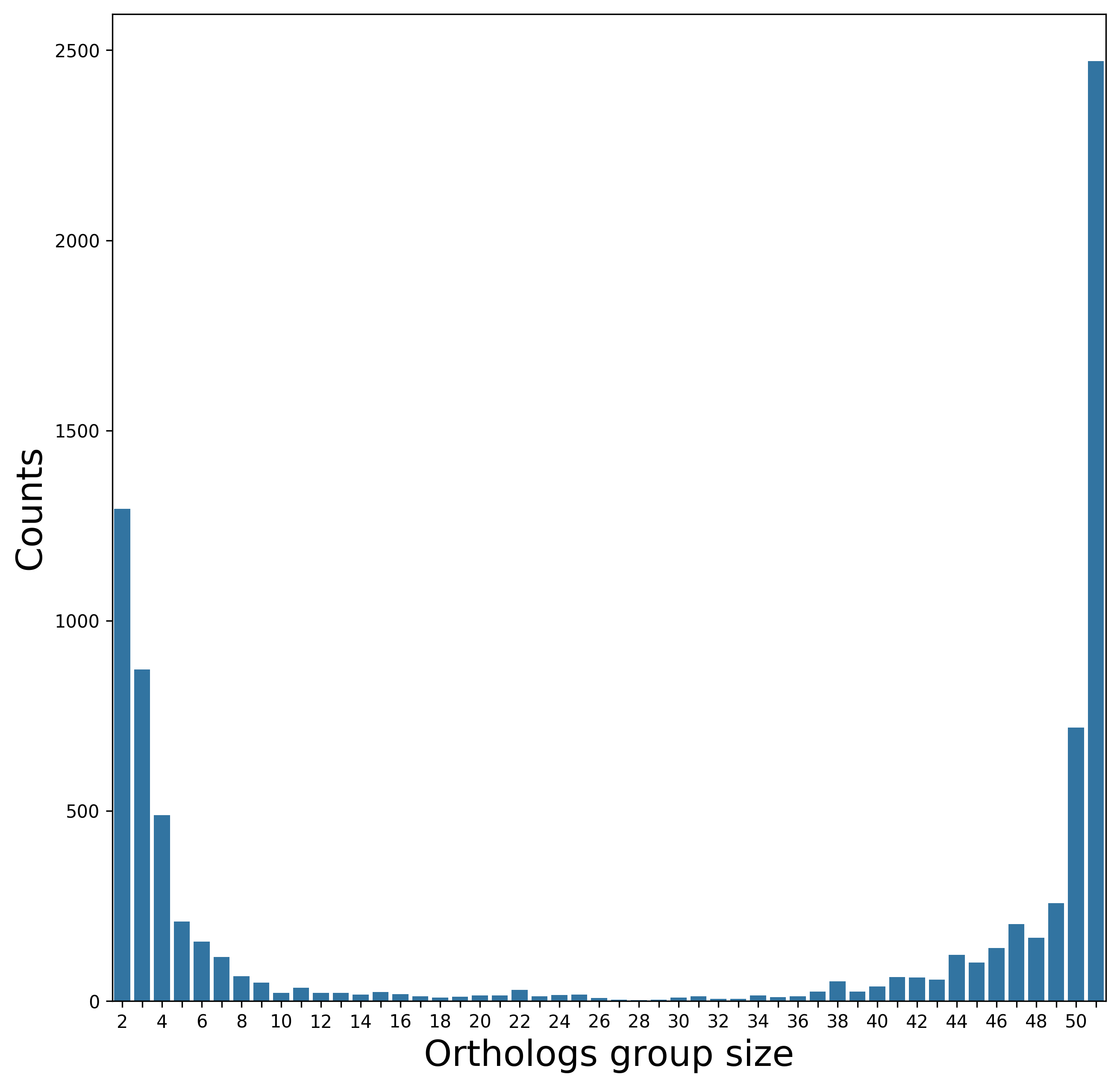
Biological interpretation: As can be seen, many genes are shared by all isolates (2,471 core genes). This is in agreement with the analyzed phylogenetic range. When closely related genomes are analyzed (isolates of the same species in this case), one expects a high number of core genes (a shift to the right side of the distribution). Indeed, when more diverged genomes were analyzed, the number of core genes was substantially lower (see other examples in this Gallery). The left side of the distribution may indicate lateral transfer events to a sub population of the analyzed genomes.
[1d] Phylogenetic tree (click on the figure to move to open the interactive mode)
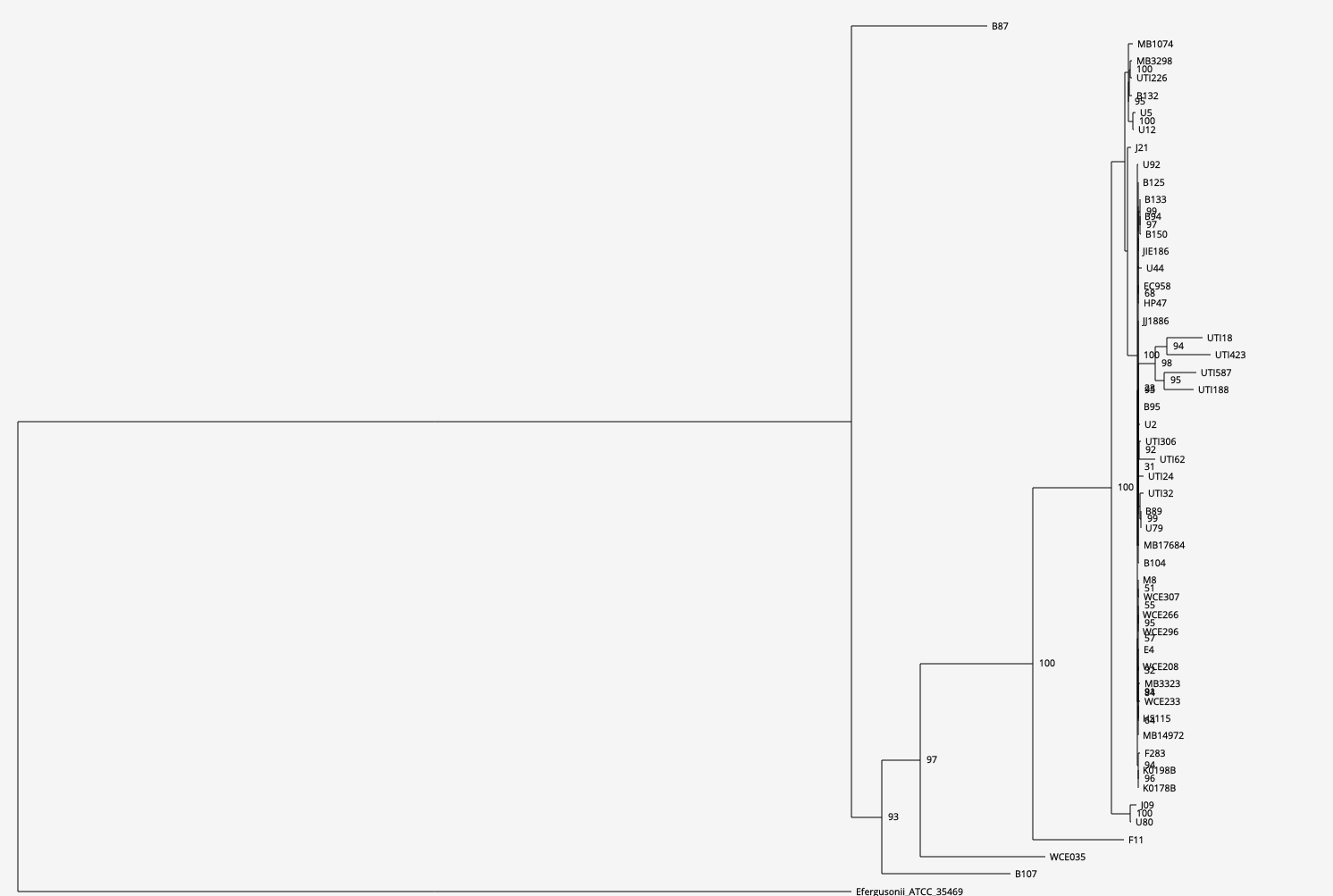
Ignoring E. fergusonii that was added for rooting, the mostly diverged genomes on the tree are isolates B107 and B87 (their divergence is evident also by the long branches leading to these genomes). In agreement with their divergence from the main group of isolates, B87 has both an atypically low number of ORFs (4,596, the second lowest) and the highest GC content (above 52%). B107 has an atypically high number ORFs (5,305, the third highest).
[2] M1CR0B1AL1Z3R analysis of different Escherichia species (including 10 Shigella). The data were downloaded from the NCBI repository, comprising of 73 genomes: 62 E. coli species, 10 Shigella (that are considered as E. coli as well) and one E. fergusonoii. The results of this example can be found here.
[2a] Below is a violin plot representing the dispersion of the ORF count per genome.
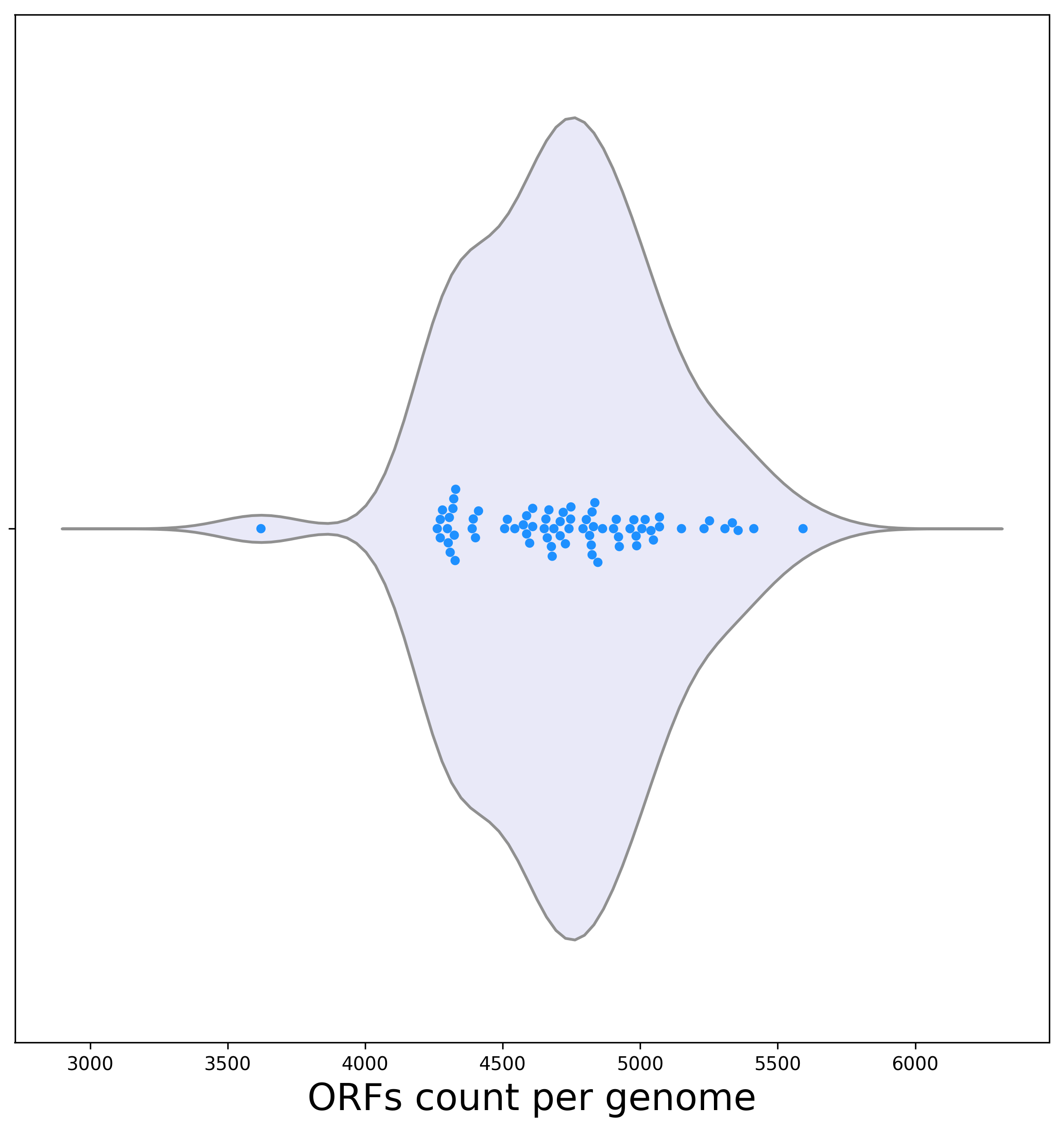
Biological interpretation: Here, one genome (E. coli K12 MDS42) had the lowest number of ORFs: 3621 in total. One possible explanation for a low number of ORFs may be missing data. However, here this is clearly not the case, as this genome is composed of one contig only, with high coverage. In this case the explanation for the low number of ORFs is that the strain was engineered using synthetic biology techniques (Posfai G, et al., 2006). It is a multiple-deletion series (MDS) strains, in which 15% of the genome was deleted, including many nonessential genes. The second smallest genome in terms of number of ORFs is E. fergusonii with 4,262 ORFs. Notably, even when this genome is excluded, genomes differ by more than 1,000 ORFs. As stated above, this can often be attributed to the high rate of horizontal gene transfer. As these genomes are highly annotated and well assembled, it is less likely that the variance is due to missing data in some of the genomes.
[2b] Below is a violin plot representing the dispersion of the GC content among genomes.
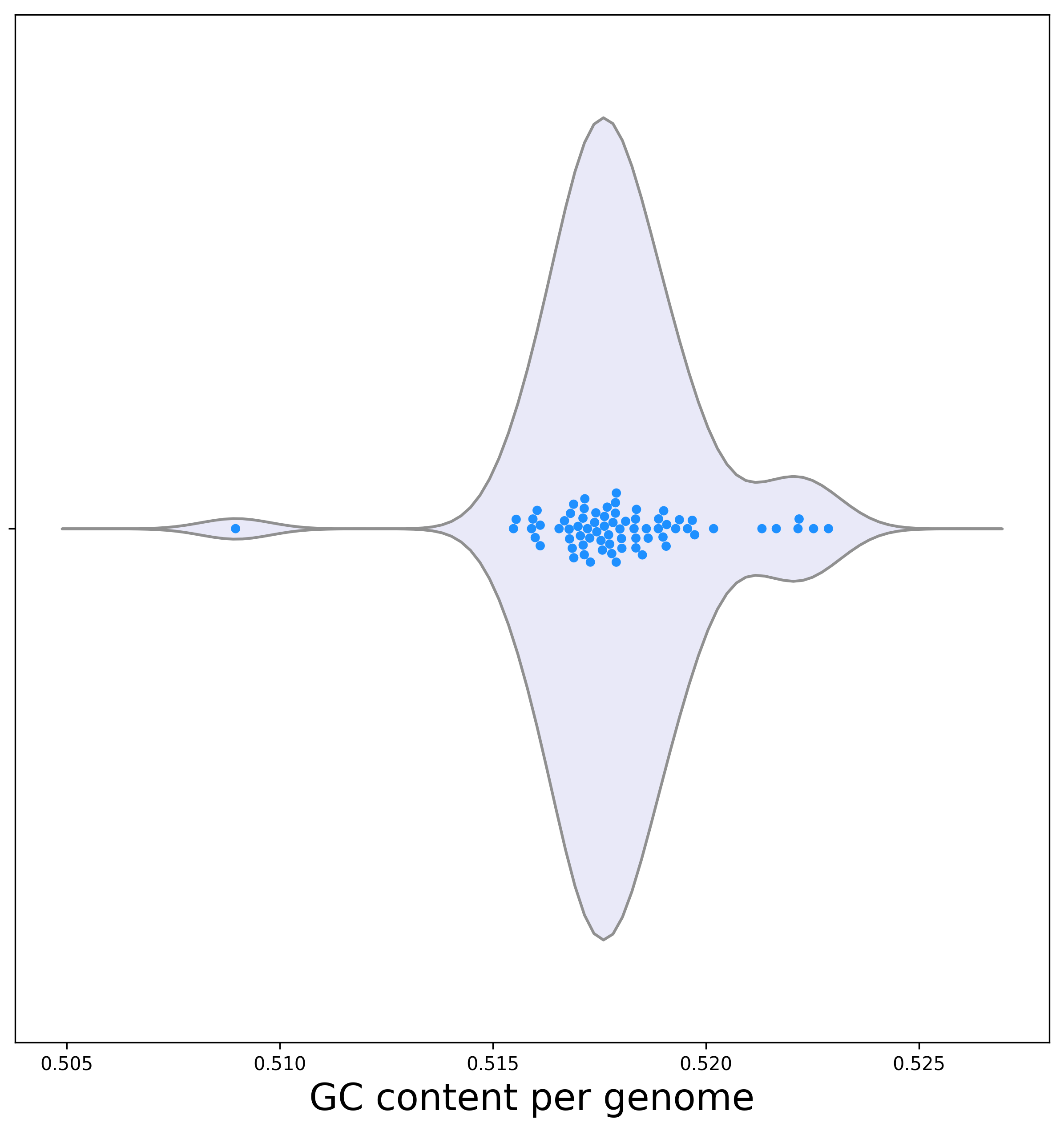
Biological interpretation: Except for one genome, the GC content is relatively uniform ranging between 51.5% and 52.5%. The genome with the atypical GC content is E. fergusonii. This is expected as GC content varies among bacterial clades and E. fergusonii is the outgroup genome, which is evolutionary diverged from all other genomes.
[2c] Orthologs detection
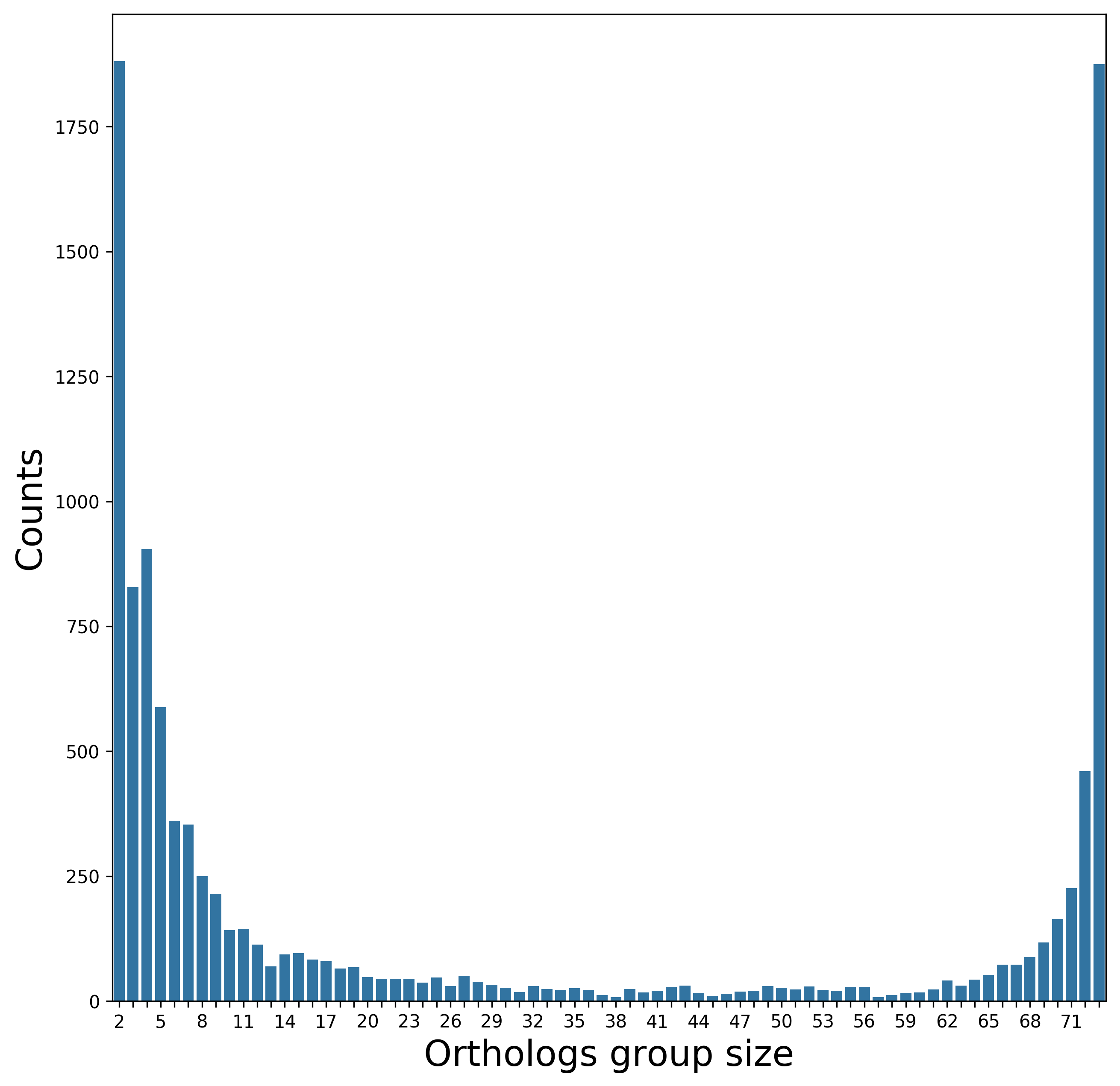
Biological interpretation: there are 1,878 core genes (right most bar), shared by all 73 strains. We speculate that some of the orthologous groups shared by 72 and 71 genomes are core genes that are absent in one or two samples because of missing data, or because they were excluded by the specific thresholds used in the orthologs identification step (see "What are the tunable parameters and how can they influence my results?").
[2d] Phylogenetic tree (click on the figure to move to open the interactive mode)
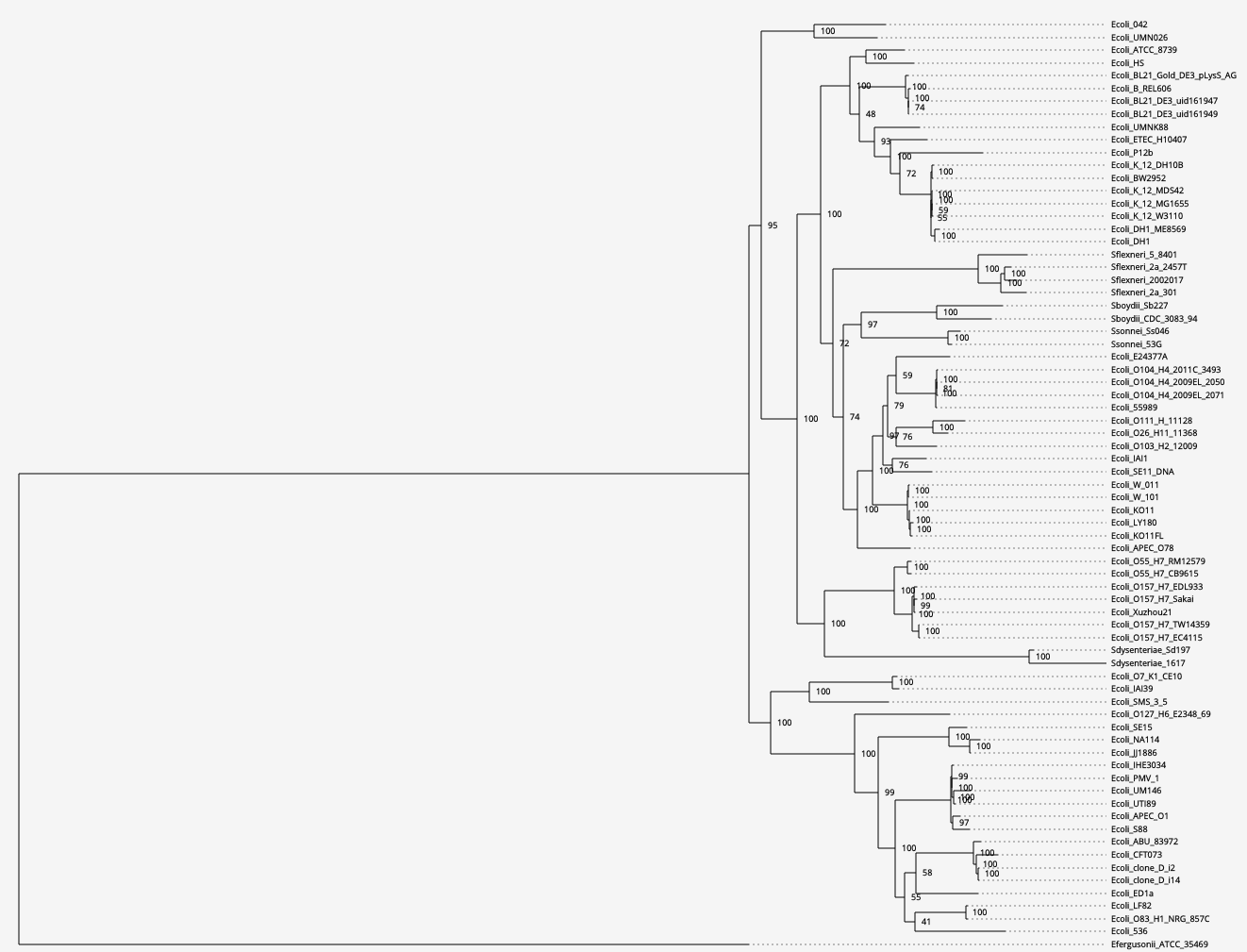
Biological interpretation: the tree is consistent with previously published E. coli trees (e.g., Oren Y, et al., 2014). As expected, the genome of E. coli K12 MDS42 clusters with MG1655 from which it originated. The large branch separating the outgroup from the ingroup genomes suggests these two species had substantial evolutionary time to diverge, in agreement with the differences in GC content between the genomes of E. fergusonii and E. coli.
[3] M1CR0B1AL1Z3R analysis of data from different species, representing the Gammaproteobacteria diversity. The data were downloaded from here. We analyzed 29 genomes. This demonstrates the capability of M1CR0B1AL1Z3R to handle highly diverged genomes. The results for this example can be found here.
[3a] Below is a violin plot representing the dispersion of the ORF count per genome. The violin plot representing the dispersion of the ORF count per genome suggests very high variance.
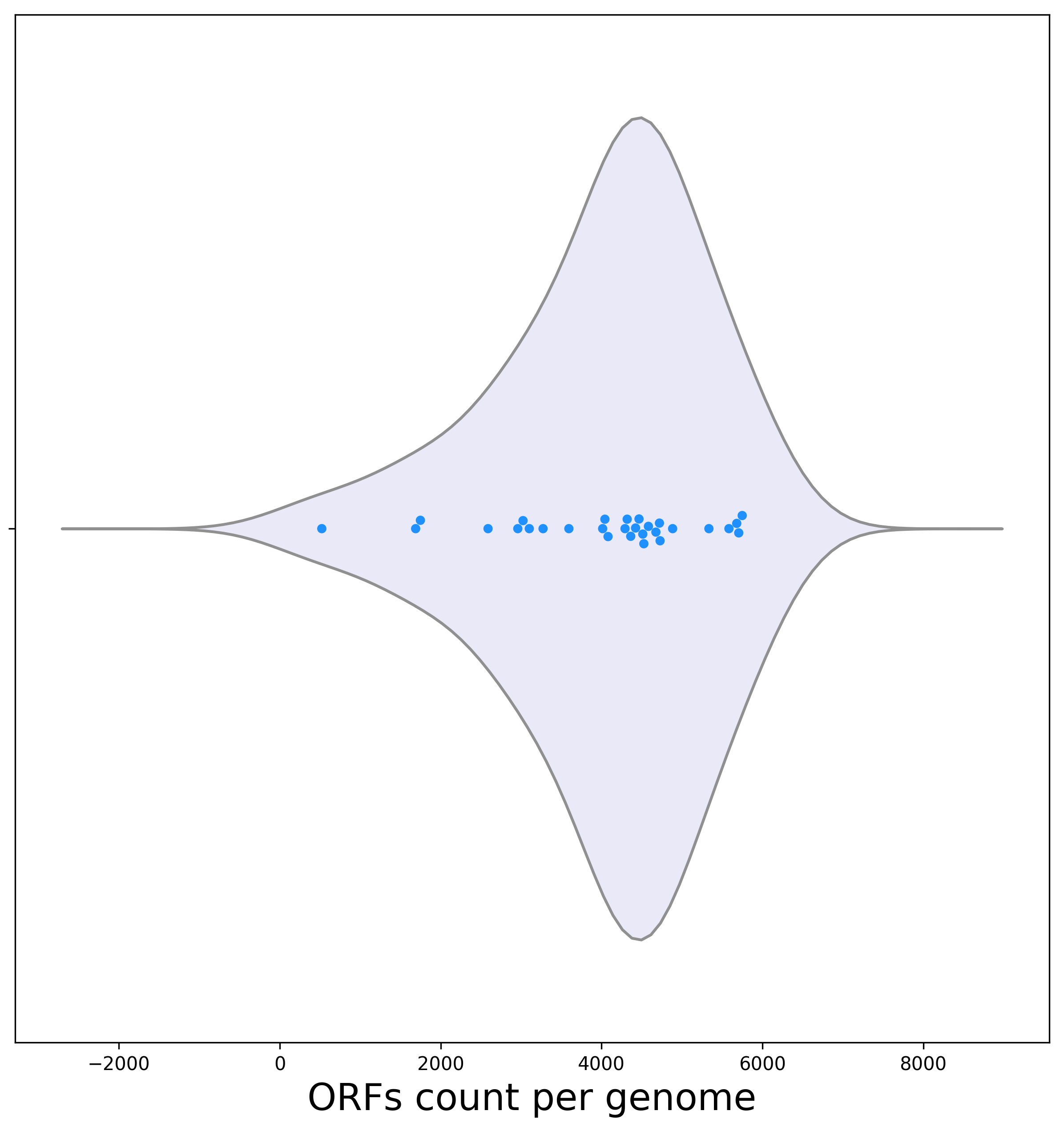
Biological interpretation: The genome of Buchnera aphidicola Bp has only 522 ORFs, while the genome Photobacterium profundum has over 10 fold more ORFs (5,749 ORFs). This high diversity is expected as we analyze phylogenetically distant bacterial clades.
[3b] The violin plot representing the GC content dispersion among genomes shows high variance as well:
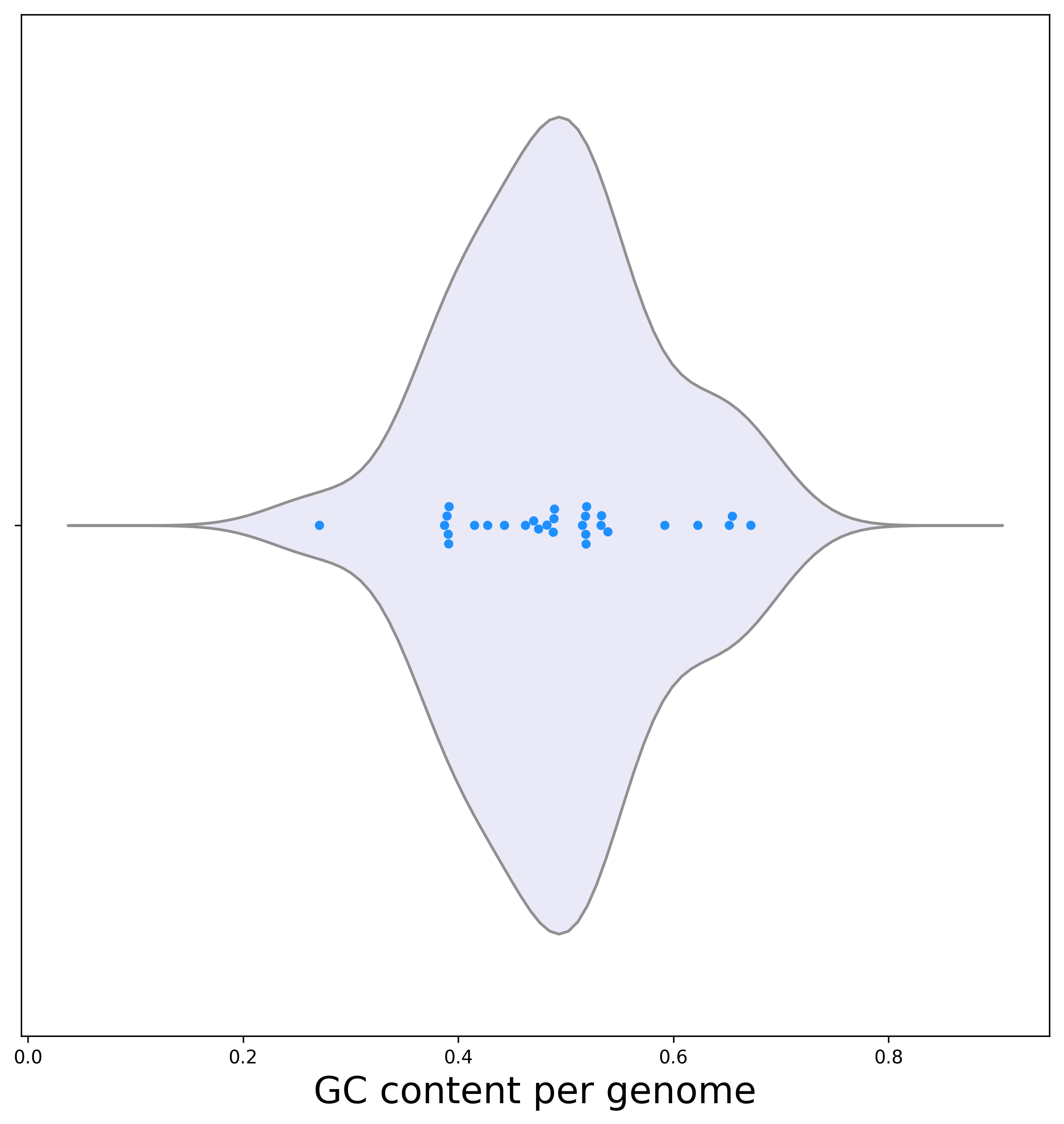
Biological interpretation: GC content is known to substantially vary among diverged groups of bacteria. Hence it is not surprising that the highest variance among the three analyzed examples is found here, where diverged groups of bacteria are analyzed. Buchnera aphidicola Bp has the lowest GC content , only 27.1%. As mentioned above, this bacterium also encodes the lowest number of ORFs. These results are consistent with this strain being an obligate endosymbiont. It was previously shown that shifts to such a life style is associated with loss of genes and a decrease in GC content (van Ham RC, et al., 2003).
[3c] Orthologs detection

Biological interpretation: Only three genes were inferred to be shared by all analyzed genomes, in line with their high divergence.
[3d] Phylogenetic tree. The tree is based on an alignment that is comprised of 51,552 amino acid sites. (click on the figure to move to open the interactive mode)
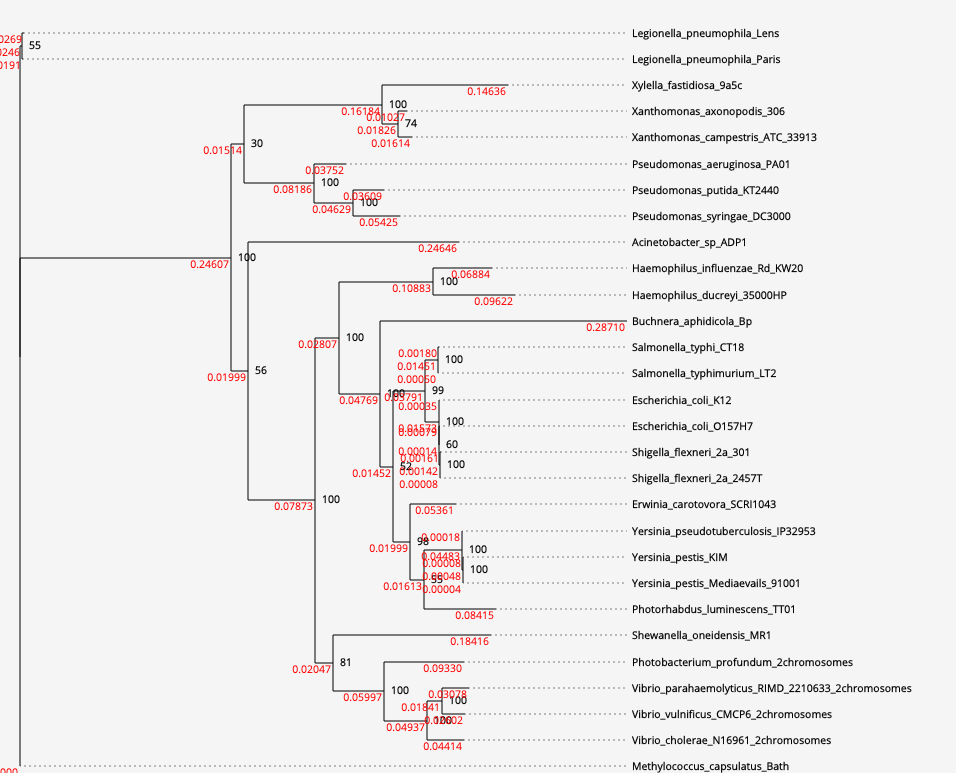
Biological interpretation: The tree reconstruction set relied on 160 "core genes", defined here as those shared by at least 50% genomes (see "What are the tunable parameters and how can they influence my results?"). This low core genes threshold was selected because only three genes are shared by all the species. Trees that are based on such a small dataset may be highly unreliable (see "Can I trust the obtained core gene phylogenetic tree?"). Indeed, when we tried reconstructing the tree based on the three shared genes only, we obtained a similar tree, albeit with much lower bootstrap values.
[1] 50 E. coli isolates and one E. fergusonii as an outgroup.
[2] 73 Escherichia species (including 10 Shigella).
[3] 29 Gammaproteobacteria species.
[2] 73 Escherichia species (including 10 Shigella).
[3] 29 Gammaproteobacteria species.
Of note, these examples highlight the main usage of the web server: a comparative analysis of assembled microbial genomes. It is not intended for raw metagenomics data (i.e., before assembly and binning). The results for each dataset are provided below.
[1] M1CR0B1AL1Z3R analysis of different isolates from the same E. coli strain. This dataset is comprised of 50 pathogenic Escherichia coli lineage ST131 genomes (McNally A, et al., 2016). This dataset represents highly similar clinical isolates of a specific bacterial species. Several isolates were not fully assembled and contained dozens and even hundreds of contigs. This demonstrates the capability of M1CR0B1AL1Z3R to handle partially assembled genomes. We added an outgroup sequence to this dataset, the genomic sequence of Escherichia fergusonii. The results for this example can be found here.
[1a] The identification of ORFs in each genome is one of the first computation steps. Below is a violin plot representing the dispersion of the ORF count per genome.
Biological interpretation: As can be seen, there is high variation in the number of encoded ORFs. This variation is often attributed to the high rate of horizontal gene transfer. However, it is also possible that some variance may result from missing data in some genomes. This plot can thus provide a gateway for further research concerning the source of differences in the number of ORFs among genomes. The genome with the lowest number of ORFs is E. fergusonii ATCC 35469. It encodes 4,262 ORFs. The next smallest genome in terms of ORF count encodes 4,495 ORFs (genome J09).
[1b] An additional analysis that is conducted before searching for orthologous sequences is the determination of the distribution of the GC composition (the relative proportion of the G and C nucleotides out of the total number of nucleotides in protein coding sequences). Below is a violin plot representing the dispersion of the GC among genomes.
Biological interpretation: the vast majority of genomes have a GC content in the range of 51% to 52%. Few variants show atypical GC content. The names of the isolates with a lower GC content can be extracted from the provided output file. For example, the isolate with the lowest GC content is UTI423. It could be interesting to further study whether a correlation exists between isolates with the atypical GC content and those with extreme number of ORFs, or if they are relatively diverged from the other isolates on the phylogenetic tree. Testing this, we found that the two genomes (UTI423 and UTI587) with the lowest GC content (less than 50%) had the highest number of ORFs. This can be explained by extensive recent events of horizontal gene transfer acquired from AT rich donors. Of note, horizontally transferred genes are often AT rich, further supporting our biological interpretation (Daubin V, et al., 2003).
[1c] Orthologs detection
Biological interpretation: As can be seen, many genes are shared by all isolates (2,471 core genes). This is in agreement with the analyzed phylogenetic range. When closely related genomes are analyzed (isolates of the same species in this case), one expects a high number of core genes (a shift to the right side of the distribution). Indeed, when more diverged genomes were analyzed, the number of core genes was substantially lower (see other examples in this Gallery). The left side of the distribution may indicate lateral transfer events to a sub population of the analyzed genomes.
[1d] Phylogenetic tree (click on the figure to move to open the interactive mode)
Ignoring E. fergusonii that was added for rooting, the mostly diverged genomes on the tree are isolates B107 and B87 (their divergence is evident also by the long branches leading to these genomes). In agreement with their divergence from the main group of isolates, B87 has both an atypically low number of ORFs (4,596, the second lowest) and the highest GC content (above 52%). B107 has an atypically high number ORFs (5,305, the third highest).
[2] M1CR0B1AL1Z3R analysis of different Escherichia species (including 10 Shigella). The data were downloaded from the NCBI repository, comprising of 73 genomes: 62 E. coli species, 10 Shigella (that are considered as E. coli as well) and one E. fergusonoii. The results of this example can be found here.
[2a] Below is a violin plot representing the dispersion of the ORF count per genome.
Biological interpretation: Here, one genome (E. coli K12 MDS42) had the lowest number of ORFs: 3621 in total. One possible explanation for a low number of ORFs may be missing data. However, here this is clearly not the case, as this genome is composed of one contig only, with high coverage. In this case the explanation for the low number of ORFs is that the strain was engineered using synthetic biology techniques (Posfai G, et al., 2006). It is a multiple-deletion series (MDS) strains, in which 15% of the genome was deleted, including many nonessential genes. The second smallest genome in terms of number of ORFs is E. fergusonii with 4,262 ORFs. Notably, even when this genome is excluded, genomes differ by more than 1,000 ORFs. As stated above, this can often be attributed to the high rate of horizontal gene transfer. As these genomes are highly annotated and well assembled, it is less likely that the variance is due to missing data in some of the genomes.
[2b] Below is a violin plot representing the dispersion of the GC content among genomes.
Biological interpretation: Except for one genome, the GC content is relatively uniform ranging between 51.5% and 52.5%. The genome with the atypical GC content is E. fergusonii. This is expected as GC content varies among bacterial clades and E. fergusonii is the outgroup genome, which is evolutionary diverged from all other genomes.
[2c] Orthologs detection
Biological interpretation: there are 1,878 core genes (right most bar), shared by all 73 strains. We speculate that some of the orthologous groups shared by 72 and 71 genomes are core genes that are absent in one or two samples because of missing data, or because they were excluded by the specific thresholds used in the orthologs identification step (see "What are the tunable parameters and how can they influence my results?").
[2d] Phylogenetic tree (click on the figure to move to open the interactive mode)
Biological interpretation: the tree is consistent with previously published E. coli trees (e.g., Oren Y, et al., 2014). As expected, the genome of E. coli K12 MDS42 clusters with MG1655 from which it originated. The large branch separating the outgroup from the ingroup genomes suggests these two species had substantial evolutionary time to diverge, in agreement with the differences in GC content between the genomes of E. fergusonii and E. coli.
[3] M1CR0B1AL1Z3R analysis of data from different species, representing the Gammaproteobacteria diversity. The data were downloaded from here. We analyzed 29 genomes. This demonstrates the capability of M1CR0B1AL1Z3R to handle highly diverged genomes. The results for this example can be found here.
[3a] Below is a violin plot representing the dispersion of the ORF count per genome. The violin plot representing the dispersion of the ORF count per genome suggests very high variance.
Biological interpretation: The genome of Buchnera aphidicola Bp has only 522 ORFs, while the genome Photobacterium profundum has over 10 fold more ORFs (5,749 ORFs). This high diversity is expected as we analyze phylogenetically distant bacterial clades.
[3b] The violin plot representing the GC content dispersion among genomes shows high variance as well:
Biological interpretation: GC content is known to substantially vary among diverged groups of bacteria. Hence it is not surprising that the highest variance among the three analyzed examples is found here, where diverged groups of bacteria are analyzed. Buchnera aphidicola Bp has the lowest GC content , only 27.1%. As mentioned above, this bacterium also encodes the lowest number of ORFs. These results are consistent with this strain being an obligate endosymbiont. It was previously shown that shifts to such a life style is associated with loss of genes and a decrease in GC content (van Ham RC, et al., 2003).
[3c] Orthologs detection
Biological interpretation: Only three genes were inferred to be shared by all analyzed genomes, in line with their high divergence.
[3d] Phylogenetic tree. The tree is based on an alignment that is comprised of 51,552 amino acid sites. (click on the figure to move to open the interactive mode)
Biological interpretation: The tree reconstruction set relied on 160 "core genes", defined here as those shared by at least 50% genomes (see "What are the tunable parameters and how can they influence my results?"). This low core genes threshold was selected because only three genes are shared by all the species. Trees that are based on such a small dataset may be highly unreliable (see "Can I trust the obtained core gene phylogenetic tree?"). Indeed, when we tried reconstructing the tree based on the three shared genes only, we obtained a similar tree, albeit with much lower bootstrap values.