evolutionary rate shifts
in sequence sites
Prof. Itay Mayrose Lab - Plant Evolution, bioinformatics, & comparative genomics
|
|
Detecting trait-dependent evolutionary rate shifts in sequence sites Prof. Itay Mayrose Lab - Plant Evolution, bioinformatics, & comparative genomics |
|
| HOME OVERVIEW GALLERY SOURCE CODE CITING & CREDITS | ||
TraitRateProp is a probabilistic method that allows testing whether the rate of sequence evolution of an examined protein or genomic region is associated with a binary phenotypic character trait. The method further allows the detection of specific sequence sites whose evolutionary rate is most noticeably affected following the character transition, suggesting a shift in functional/structural constraints.
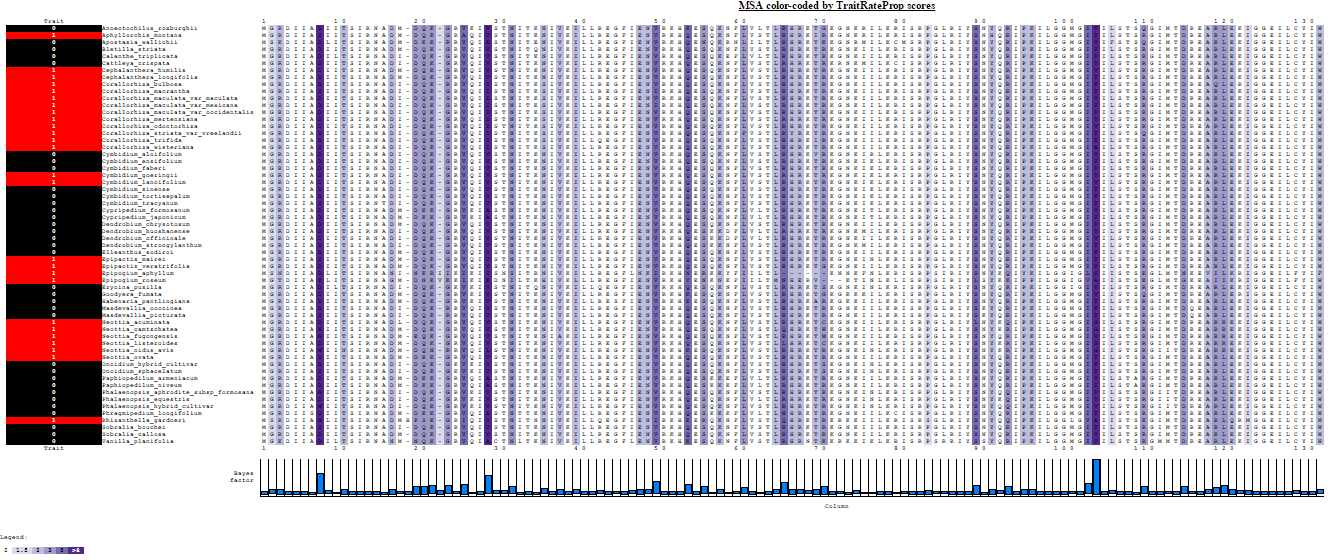
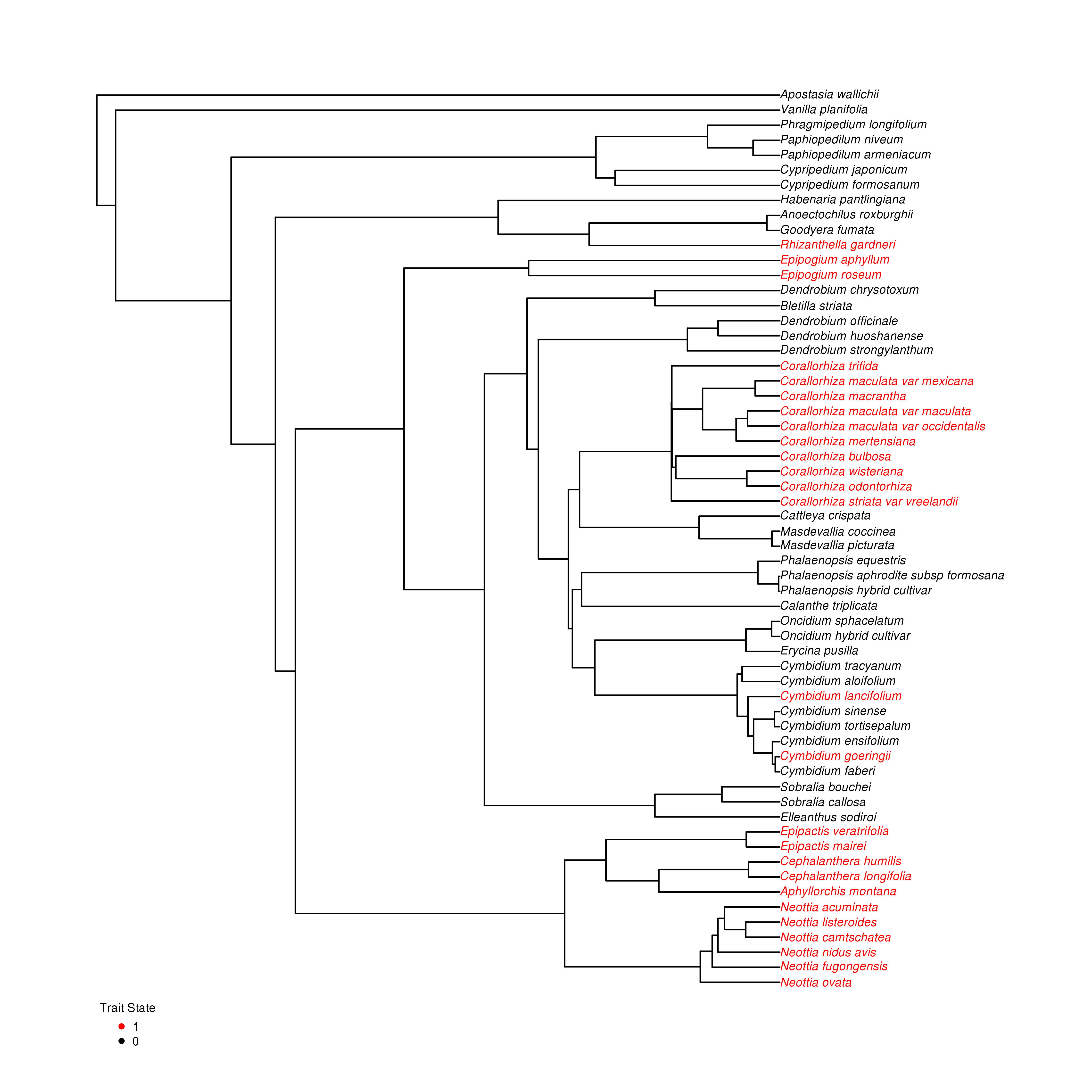
TraitRateProp detects cases in which some or all sequence positions in a given gene (protein) exhibit evolutionary rate shifts that are associated with the state of a binary phenotypic trait. The trait can be related to a genomic attribute (e.g., the presence/absence of a certain gene family) or to an organismal trait (e.g., an environmental or ecological preference, life history attribute, or morphological feature). Given an input rooted ultrametric species tree, a multiple sequence alignment (MSA), and the characters describing the trait states of the extant species (coded as either '0' or '1'), TraitRateProp allows for: (1) testing whether the evolutionary rate of the input sequence data is associated with the given trait data; (2) In case an association is detected, the method infers the sequence positions whose evolutionary rate is most likely to be associated with the trait data. TraitRateProp is based on the maximum-likelihood paradigm, and provides two important maximum likelihood estimators (MLEs) regarding the co-evolution of sequence and trait data: the relative rate parameter, r, describing the ratio between the sequence evolutionary rates under states '1' and '0', and the parameter, p, which is the proportion of positions in the sequence whose evolutionary rate is associated with the phenotypic state. The full details of the model, the likelihood estimation procedures and the associated statistical tests are detailed in (Levy Karin et al.; Mayrose and Otto).
TraitRateProp combines models of sequence evolution and of phenotypic trait evolution in a single likelihood framework by first reconstructing a large number of possible evolutionary histories of the phenotypic trait along the phylogeny. Each such history is inferred using the stochastic mapping approach (Nielsen) and is consistent with the observed phenotypic state values of the extant species. The method is based on comparing a null model, in which a single sequence rate matrix is fit to the data and an alternative model, in which two sequence rate matrices, each corresponding to one of the phenotypic states, are fit to the sequence data.
Connecting the trait and sequence evolutionary processes:
Dependence of the rate of sequence evolution on the phenotypic state is modelled by allowing the sequence evolutionary rate of some positions termed "phenotype-dependent positions" to vary depending on whether the phenotypic trait is in state '0' or '1'. Specifically, a parameter r1 is assumed when the character state is '1', and a parameter r0 when the character state is '0'. Thus, for a phenotype-dependent position, the sequence rate matrix, is multiplied by either r1 or r0, according to the character state. The parameter r denotes the ratio between r1 and r0.
The TraitRateProp joint genotype-phenotype likelihood framework describes an evolutionary process along a phylogeny, and considers two types of data: sequence data (DS) and character states (DC) of the extant species. The likelihood of the model is the joint probability of DS and DC given the model parameters θ. This expression is termed as the probability to observe DC times the probability to observe DS conditioned on having observed DC. Under these settings, likelihood computations based on the sequence data require the knowledge of the character state in each part of T, i.e., the complete reconstructed history of character changes. As this history is unknown the marginal probability of DS given DC and θ is approximated by integrating over many possible character histories h (as obtained using the stochastic mappings approach).
The likelihood based on each sequence position k is computed using a mixture model of the likelihoods over two scenarios: either the position evolved independently of the character state or the position belongs to the phenotype-dependent category. In this mixture model, the parameter p specifies the probability of a position to belong to the phenotype-dependent category. By fixing p to 1, the user can test the hypothesis that the evolutionary rate of all sequence sites is associated with the examined trait.
A rooted ultrametric phylogentic tree with branch lengths (Newick format).
A multiple sequence alignment (MSA) of the sequence data of the extant species (Fasta format).
The character states of the extant species coded as either '0' or '1' (Fasta format).
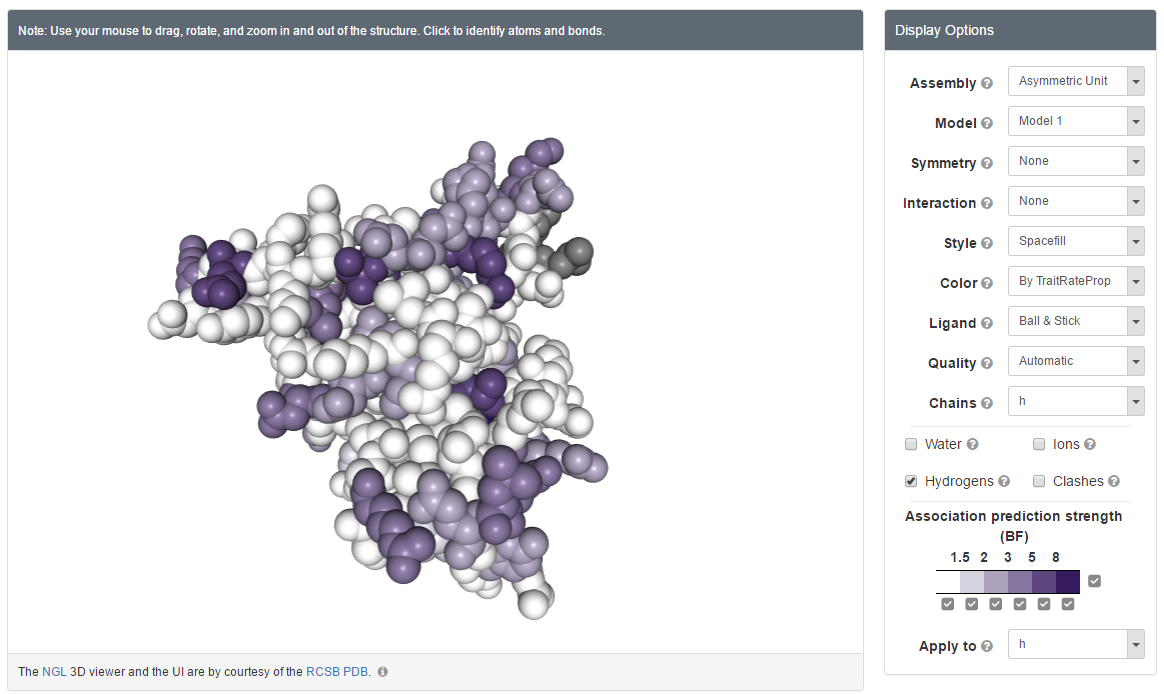
In addition, the user should indicate the type of sequence input (DNA or protein). The user can also control the search range of the r parameter and whether the p parameter should be optimized or not. Fixing the p parameter to 1 allows the user to run the program in TraitRate mode, assuming that the evolutionary rate of all sequence sites is associated with the examined trait. Finally, in case of protein data, the user can provide a 3D structural model in the form of a PDB file format. In this case, the site-specific predictions of TraitRateProp are projected onto the provided 3D protein structure.
TraitRateProp directs you to a web page called "TraitRateProp Job Status Page". This web page is automatically updated every 30 seconds, showing messages regarding the different stages of the server activity.
A runtime estimation is computed based on the size of the provided input. A basic linear regression model through the origin was pre-computed from simulated datasets that contained 1,000 sequence position (MSA length) with a varying number of species:
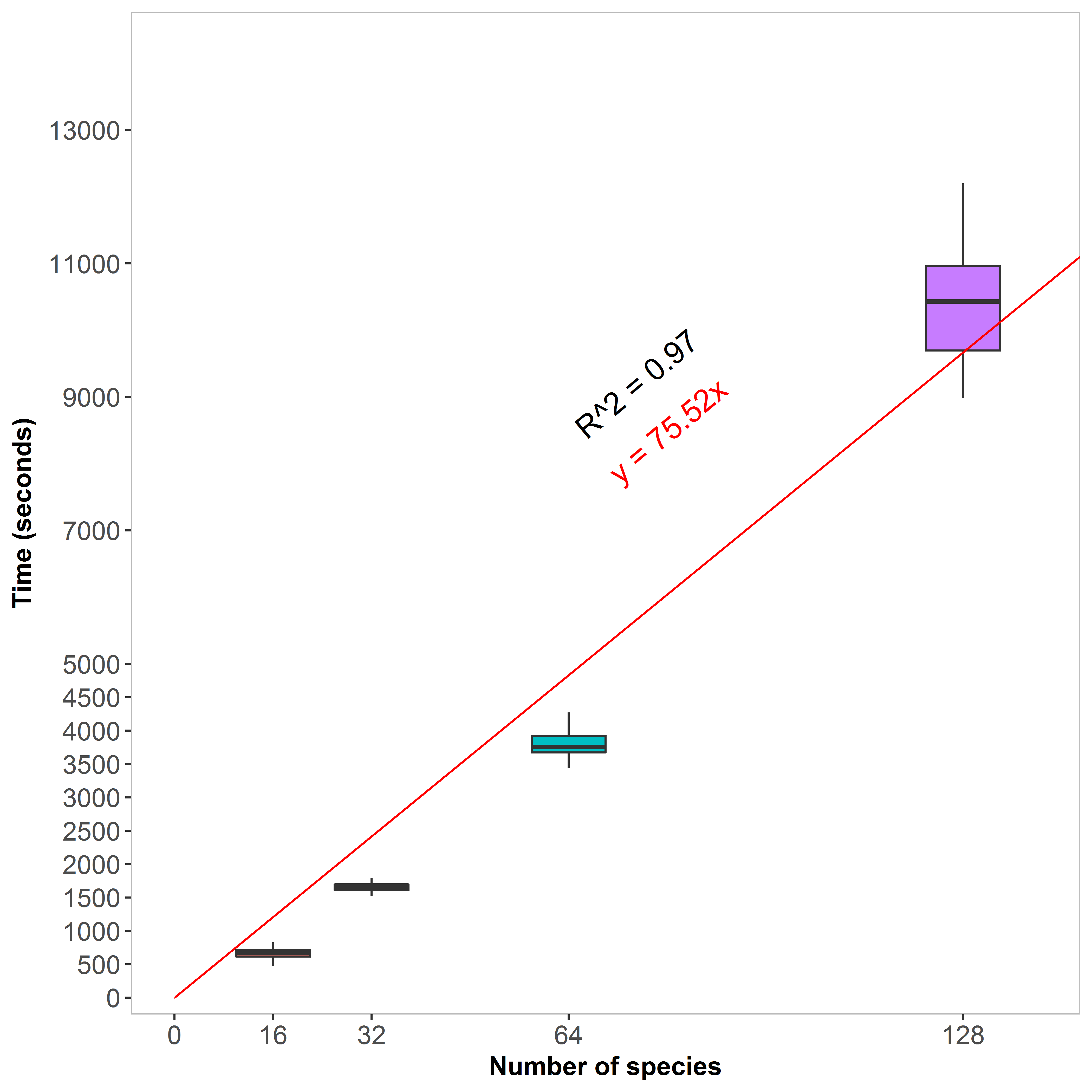
As the runtime is expected to increase linearly with the number of species (N) and the number of sequence positions (L), the TraitRateProp web server uses the following formula to estimate the runtime in seconds:
Parameter estimations and model comparison: a result of a chi-squared hypothesis testing to compare the null model (no association between shifts in the phenotypic trait and the rate of sequence evolution) and the alternative model are printed to the result page together with the alternative model's r and p estimations. A full report of the parameter estimations under the null and alternative models and the comparison between them is provided in a downloadable file